
Digital twins

Decentralized patients recruitment

Real-World Data

Passive Monitoring

Automated patient engagement
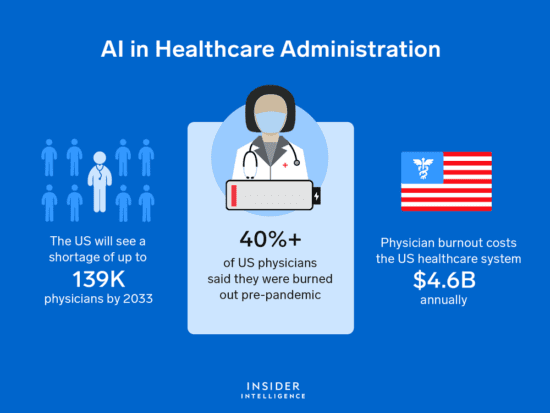
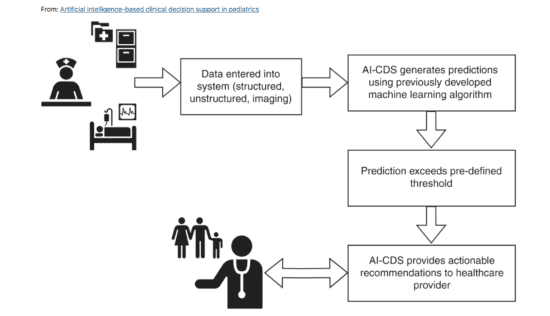



Zoi Capital | Digital Health – AI in Healthcare – Venture Capital
